The Therapeutic Cancer Cell Atlas
Welcome to TCCA
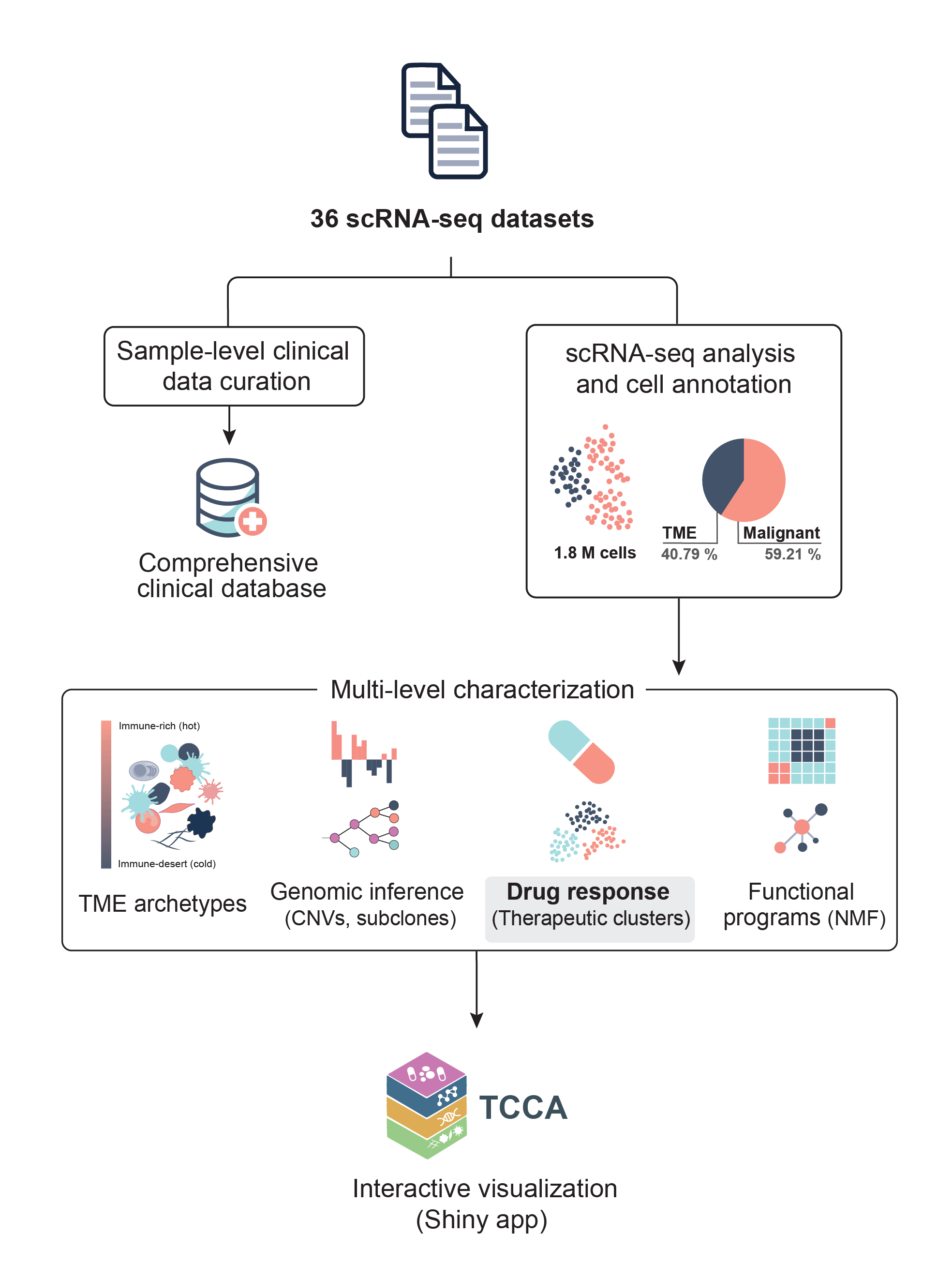
The Therapeutic Cancer Cell Atlas (TCCA) is a comprehensive resource designed to explore how intra-tumoral heterogeneity (ITH) influences drug sensitivity across cancer types, providing insights into treatment responses and potential therapeutic targets.
TCCA integrates single-cell RNA sequencing (scRNA-seq) data from 36 public studies, encompassing 1.8 million single-cell transcriptomes across 34 cancer types, including 670 samples from 540 patients and 183 samples from cell lines. This pan-cancer cohort enables systematic exploration of transcriptional ITH and its therapeutic implications.
Key features of TCCA:
- Multi-layer molecular integration: Combines genetic copy number alteration (CNA) profiles, functional metaprograms, and tumor microenvironment (TME) composition with therapeutic predictions.
- Drug sensitivity predictions: Leverages methodologies like Beyondcell and scTherapy to predict drug responses of genetically distinct subclones based on their transcriptional profiles.
- Functional characterization: Systematically identifies recurrent gene modules using Non-Negative Matrix Factorization (NMF) and links them to drug-specific vulnerabilities.
- TME influence analysis: Examines how distinct TME archetypes affect drug responses across different cancer contexts.
- Pan-cancer therapeutic clusters: Identifies 10 recurrent clusters, including subclones from diverse tumors sharing aggressive phenotypes and potential therapeutic vulnerabilities.
- Patient stratification tools: Interactive visualizations and downloadable reports to guide experimental validation of drug sensitivity predictions from single-cell tumor profiles, ultimately supporting clinical decision-making.
TCCA is developed and maintained by the CNIO Bioinformatics Unit:
- CNIO Bioinformatics Unit: Supporting CNIO research and developing computational methods for cancer genomics, integrating multi-omics data to advance precision oncology.
Disclaimer: This resource is exclusively intended for research purposes and academic use. It should not be used for medical or professional advice.
Please Cite Us:
- González-Bermejo, M. et al. (2025). Revealing the single-cell therapeutic landscape across cancers [Manuscript in preparation].
Follow us:
Genomic vs Therapeutic heterogeneity
Each point represents a sample. Use the panels below to analyze a single patient or a clinical cohort.
Search patient
Select a patient cohort by clinical variables
Select one or more values per variable to define a cohort. All patients matching these criteria will be included.
Predicted drugs across selected samples
Subclones across selected samples
Sample details
No data matches the current selection
Select a patient or adjust cohort filters.
TME distribution across clinical variables:
TCCA dataset repository
Comprehensive single-cell therapeutic atlas across cancers. All data are hosted on Zenodo and available for download.
Main single-cell atlas (.h5ad) containing raw and normalized counts, embeddings, cell metadata, TME archetypes, subclones, and therapeutic clusters. Cell-level metadata (TSV) is also available for simplified downstream analyses.
Comprehensive metadata for all 36 source studies, including study identifiers, GEO accessions, tumor types, sample and cell counts, and PMIDs.
Clinical annotations per sample including sex, age, cancer type, tumor site, sample type, and treatment status.
Proportions of 12 immune and stromal cell types per sample, along with TME archetype classification of samples based on unsupervised clustering.
Copy number variation profiles per subclone and cell-level clonality annotations inferred using SCEVAN.
Predicted drug responses for each SCEVAN-inferred subclone with scTherapy, Beyondcell drug sensitivity scores per subclone, therapeutic cluster gene signatures
NMF-derived transcriptional programs, functional enrichment analysis results, and UCell scores for malignant cells.
Complete computational pipeline and source code used to generate the Therapeutic Cancer Cell Atlas (TCCA) and all downstream analyses.
About TCCA
Unveiling therapeutic heterogeneity
Intratumoral heterogeneity (ITH) is a major determinant of therapeutic failure, yet its impact on drug response across cancers remains incompletely understood. The Therapeutic Cancer Cell Atlas (TCCA) addresses this challenge as a pan-cancer single-cell resource that integrates approximately 1.8 million transcriptomes from 540 patients and 183 cancer cell lines spanning 34 tumor types.
By combining single-cell transcriptomics with copy-number alteration (CNA) inference and computational drug-response prediction, TCCA systematically maps therapeutic heterogeneity at subclonal resolution. This multidimensional approach allows for the identification of drug vulnerabilities that are often masked in bulk sequencing data.
Beyond genomic diversity
A core finding of this resource is that therapeutic heterogeneity is largely decoupled from genomic and transcriptomic diversity . Instead, drug sensitivity arises from distinct functional transcriptional programs and tumor microenvironment (TME) states. TCCA integrates transcriptional metaprograms and TME archetypes to reveal how stress responses, proliferative states, lineage programs, and immune context shape drug sensitivity beyond the tissue of origin.
Through this framework, we have identified ten recurrent therapeutic clusters that capture conserved and context-specific drug vulnerabilities across tumor lineages. These clusters group subclones from diverse tumors that share aggressive phenotypes and potentially actionable therapeutic targets.
Translational impact
The translational relevance of TCCA is demonstrated by linking these therapeutic clusters to patient outcomes and validating predicted vulnerabilities using independent pharmacogenomic datasets. This includes clinically actionable examples in aggressive tumor subtypes, offering a scalable framework to guide therapeutic prioritization, drug repurposing, and combination strategies in precision oncology.
Resource statistics
1.8M
Single-cells
34
Cancer types
853
Tumor samples
36
Studies
537
Unique patients
183
Cell Lines
About the CNIO Bioinformatics Unit
TCCA has been designed, created, and is maintained by the Bioinformatics Unit of the Spanish National Cancer Research Centre (BU-CNIO) .
Our mission is to support CNIO research groups and conduct original research in bioinformatics, focusing on the development of novel computational methods for integrating cancer genomics data with clinical and pathological information. Our final goal is to translate this knowledge into effective and personalized cancer treatments.
Disclaimer: This resource is exclusively intended for research purposes and academic use. It should not be used for medical or professional advice.
Please Cite Us:
- González-Bermejo, M. et al. (2025). Revealing the single-cell therapeutic landscape across cancers [Manuscript in preparation].